Medical History:
A 24-year-old female patient presented to our clinic complaining of decreased vision in both eyes.
She did not report a familiar history of retinal diseases.
Examination Findings
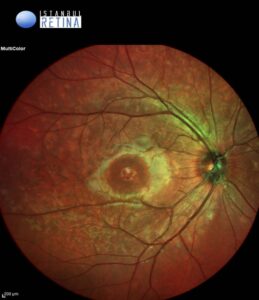
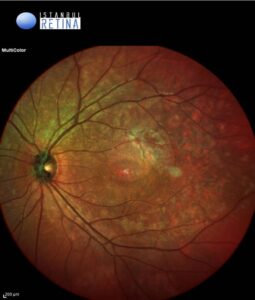
Best corrected visual acuity was 3/10 in the right eye and 3/10 in the left eye. Intraocular pressure was 16 mmHg in both eyes. Anterior segment examination was normal in both eyes. Funduscopic examination revealed bilateral multifocal vitelliform lesions (Figure 1).
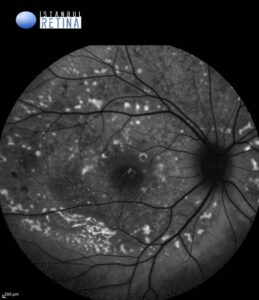
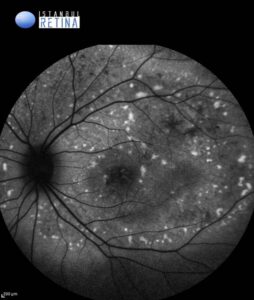
Fundus autofluorescence (FAF) imaging revealed intense hyperautofluorescence corresponding to the vitelliform lesions. (Figure 2).
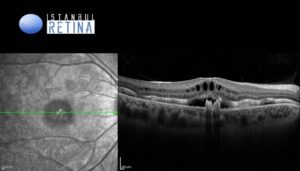
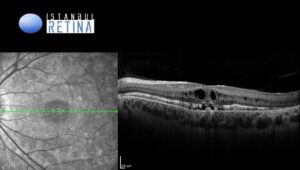
Spectral-domain optical coherence tomography (SD-OCT) showed subretinal hyperreflective deposits, cystoid maculopathy accompanied by a shallow neurosensory detachment, and outer retinal layer thickening in both eyes (Figure 3).
Diagnosis
Cystoid Maculopathy in Autosomal Recessive Bestrophinopathy
Autosomal recessive bestrophinopathy (ARB) is a hereditary retinal disorder resulting from biallelic mutations in the BEST1 gene. While phenotypic severity varies, the condition consistently manifests core features of the bestrophinopathy spectrum. A hallmark of ARB is the accumulation of both intraretinal and subretinal fluid within the macula. On OCT, the yellowish deposits observed clinically appear as hyperreflective subretinal material; however, the most striking feature is cystoid maculopathy associated with a shallow neurosensory detachment. Notably, fluorescein angiography (FA) shows no evidence of dye leakage or petaloid pooling, confirming that the cystoid spaces are non-vasogenic in nature. It remains unclear whether the presence of intraretinal fluid signifies disease progression or is a unique manifestation of specific BEST1 genotypes.
Differential Diagnosis
Multifocal pattern dystrophy, Best vitelliform macular dystrophy, adult-onset foveomacular vitelliform dystrophy, late-onset Stargardt disease
Treatment
Currently, there is no definitive cure or medical treatment to prevent the progression of autosomal recessive bestrophinopathy. Management primarily focuses on regular clinical monitoring using OCT and FAF to track the transition through the different vitelliform stages. For patients with significant central vision loss, low-vision aids and high-contrast digital devices are recommended to maintain independence. Emerging research into gene therapy remains a promising area for future treatment.
References:
Sayman Muslubas I, Arf S, Hocaoglu M, Giray Ersoz M, Karacorlu M. Best disease presenting as subretinal pigment epithelium hyperreflectivite lesion on spectral-domain optical coherence tomography: Multimodal imaging features. Eur J Ophthalmol. 2022;32:2702-2711.https://pubmed.ncbi.nlm.nih.gov/34806463/
Bianco L, Arrigo A, Antropoli A, et al. Non-vasogenic cystoid maculopathy in autosomal recessive bestrophinopathy: novel insights from NIR-FAF and OCTA imaging. Ophthalmic Genet. 2024;45:44-50.https://pubmed.ncbi.nlm.nih.gov/37041716/
Casalino G, Khan KN, Armengol M, et al. Autosomal Recessive Bestrophinopathy: Clinical Features, Natural History, and Genetic Findings in Preparation for Clinical Trials. Ophthalmology. 2021;128:706-718.https://pubmed.ncbi.nlm.nih.gov/33039401/
{kind=link}